Extracting the weighted means of individual fiber pathways can be useful when you want to quantify the microstructure of an entire white matter structure. This is specifically useful for tract based analyses, where you run statistics on specific pathways, and not the whole brain. You can read more on the distinction between tract based and voxel based analyses
here.
The prerequisite steps to get to tract based analyses are described in the tutorials on this website. In the
first tutorial we covered how to processed raw diffusion images and calculate tensor images. In the
second tutorial we described how to normalize a set of diffusion tensor images (DTI) and run statistics on the normalized brain images (including voxel bases analyses). In the
last tutorial we demonstrated who to iteratively delineated fiber pathways of interest using anatomically defined waypoints.
Here we will demonstrate and provide code examples on how to calculate a weighted mean scalar value for entire white matter tracts. The principle relies on using the density of tracts running through each voxel as a proportion of the total number of tracts in the volume to get a weighted estimate. Once you have a proportional index map for each fiber pathway of interest you can multiply this weighting factor by the value of the diffusion measure (e.g. FA) in that voxel, to get the weighted scalar value of each voxel. Shout out to Dr. Dan Grupe, who initiated and wrote the core of the weighted mean script. As a note, this script can also be used to extract cluster significance from voxel-wise statistical maps. See an example of this usage at the end of this post.
The weighted mean approach allows for differential weighting of voxels within a white matter pathway that have a higher fiber count, which is most frequently observed in areas more central to the white matter tract of interest. At the same time this method will down-weigh voxels at the periphery of the tracts, areas that are often suffering from partial voluming issues, as voxels that contain white matter also overlap gray matter and/or cerebrospinal fluid (CSF).
To start off you will need a NifTI-format tract file, for example as can be exported from Trackvis. See more details on how to do this in
Tutorial 3. You also need scalars, like FA or MD files. Overview of software packages used in this code:
TrackVis by MGH
(download TrackVis here)http://fsl.fmrib.ox.ac.uk/fsl/fslwiki/FslutilsSave the weighted mean code below into a text file named "weighted_mean.sh". Make sure the file permissions for this program are set to executable by running this line after saving.
chmod 770 weighted_mean.sh
Note that the code in 'weighted_mean.sh" assumes:
- - A base directory where all folder are located with data: ${baseDir}
- - A text file with the structures you want to run. Here again the naming is defined by what the name of the file is, and that it is located in the main directory, in a folder called STRUCTURES: ${baseDir}/STRUCTURES/${region}
- - A text file with the scalars you want to run. The naming here is defined by how your scalar files are appended. E.g. "subj_fa.nii.gz"; in this case "fa" is the identifier of the scalar file: ${scalar_dir}/*${sub}*${scalar}.nii*
- - The location of all the scalar files in the scalar directory: ${scalar_dir}
- - A list of subject prefixes that you want to run.
Weighted mean code:
#!/bin/bash
# 2013-2018
# Dan Grupe & Do Tromp
if [ $# -lt 3 ]
then
echo
echo ERROR, not enough input variables
echo
echo Create weighted mean for multiple subjects, for multiple structures, for multiple scalars;
echo Usage:
echo sh weighted_mean.sh {process_dir} {structures_text_file} {scalars_text_file} {scalar_dir} {subjects}
echo eg:
echo
echo weighted_mean.sh /Volumes/Vol/processed_DTI/ structures_all.txt scalars_all.txt /Volumes/etc S01 S02
echo
else
baseDir=$1
echo "Output directory "$baseDir
structures=`cat $2`
echo "Structures to be run "$structures
scalars=`cat $3`
echo "Scalars to be run "$scalars
scalar_dir=$4
echo "Directory with scalars "$scalar_dir
cd ${baseDir}
mkdir -p -v ${baseDir}/weighted_scalars
finalLoc=${baseDir}/weighted_scalars
shift 4
subject=$*
echo
echo ~~~Create Weighted Mean~~~;
for sub in ${subject};
do
cd ${baseDir};
for region in ${structures};
do
img=${baseDir}/STRUCTURES/${region};
final_img=${finalLoc}/${region}_weighted;
for scalar in ${scalars};
do
#if [ ! -f ${final_img}_${sub}_${scalar}.nii.gz ];
#then
scalar_image=${scalar_dir}/*${sub}*${scalar}.nii*
#~~Calculate voxelwise weighting factor (number of tracks passing through voxel)/(total number of tracks passing through all voxels)~~
#~~First calculate total number of tracks - roundabout method because there is no 'sum' feature in fslstats~~
echo
echo ~Subject: ${sub}, Region: ${region}, Scalar: ${scalar}~
totalVolume=`fslstats ${img} -V | awk '{ print $1 }'`;
echo avgDensity=`fslstats ${img} -M`;
avgDensity=`fslstats ${img} -M`;
echo totalTracksFloat=`echo "$totalVolume * $avgDensity" | bc`;
totalTracksFloat=`echo "$totalVolume * $avgDensity" | bc`;
echo totalTracks=${totalTracksFloat/.*};
totalTracks=${totalTracksFloat/.*};
#~~Then divide number of tracks passing through each voxel by total number of tracks to get voxelwise weighting factor~~
echo fslmaths ${img} -div ${totalTracks} ${final_img};
fslmaths ${img} -div ${totalTracks} ${final_img};
#~~Multiply weighting factor by scalar of each voxel to get the weighted scalar value of each voxel~~
echo fslmaths ${final_img} -mul ${scalar_image} -mul 10000 ${final_img}_${sub}_${scalar};
fslmaths ${final_img} -mul ${scalar_image} -mul 10000 ${final_img}_${sub}_${scalar};
#else
# echo "${region} already completed for subject ${sub}";
#fi;
#~~Sum together these weighted scalar values for each voxel in the region~~
#~~Again, roundabout method because no 'sum' feature~~
echo totalVolume=`fslstats ${img} -V | awk '{ print $1 }'`;
totalVolume=`fslstats ${img} -V | awk '{ print $1 }'`;
echo avgWeightedScalar=`fslstats ${final_img}_${sub}_${scalar} -M`;
avgWeightedScalar=`fslstats ${final_img}_${sub}_${scalar} -M`;
value=`echo "${totalVolume} * ${avgWeightedScalar}"|bc`;
echo ${sub}, ${region}, ${scalar}, ${value} >> ${final_img}_output.txt;
echo ${sub}, ${region}, ${scalar}, ${value};
#~~ Remember to divide final output by 10000 ~~
#~~ and tr also by 3 ~~
rm -f ${final_img}_${sub}_${scalar}*.nii.gz
done;
done;
done;
fi
Once the weighted mean program is saved in a file you can start running code to run groups of subjects. See for example the script below.
Run Weighted Mean for group of subjects:
#Calculate weighted means:
echo fa tr ad rd > scalars_all.txt
echo CING_L CING_R UNC_L UNC_R > structures_all.txt
sh /Volumes/Vol/processed_DTI/SCRIPTS/weighted_mean.sh /Volumes/Vol/processed_DTI/ structures_all.txt scalars_all.txt /Volumes/Vol/processed_DTI/scalars S01 S02 S03 S04;
Once that finishes running you can organize the output data, and divide the output values by 10000. This is necessary because to make sure the output values in the weighted mean code have sufficient number of decimals they are multiplied by 10000. Furthermore, this code will also divide trace (TR) values by 3 to get the appropriate value of mean diffusivity (MD = TR/3).
Organize output data:
cd /Volumes/Vol/processed_DTI/weighted_scalars
for scalar in fa tr ad rd;
do
for structure in CING_L CING_R UNC_L UNC_R;
do
rm -f ${structure}_${scalar}_merge.txt;
echo "Subject">>subject${scalar}${structure}.txt;
echo ${structure}_${scalar} >> ${structure}_${scalar}_merge.txt;
for subject in S01 S02 S03 S04;
do
echo ${subject}>>subject${scalar}${structure}.txt;
if [ "${scalar}" == "tr" ]
then
var=`cat *_weighted_output.txt | grep ${subject}|grep ${structure}|grep ${scalar}|awk 'BEGIN{FS=""}{print $4}'`;
value=`bc <<<"scale=8; $var / 30000"`
echo $value >> ${structure}_${scalar}_merge.txt;
else
var=`cat *_weighted_output.txt | grep ${subject}|grep ${structure}|grep ${scalar}|awk 'BEGIN{FS=""}{print $4}'`;
value=`bc <<<"scale=8; $var / 10000"`
echo $value >> ${structure}_${scalar}_merge.txt;
fi
done
mv subject${scalar}${structure}.txt subject.txt;
cat ${structure}_${scalar}_merge.txt;
done
done
#Print data to text file and screen
rm all_weighted_output_organized.txt
paste subject.txt *_merge.txt > all_weighted_output_organized.txt
cat all_weighted_output_organized.txt
This should provide you with a text file with columns for each structure & scalar combination, with rows for each subject. You can then export this to your favorite statistical processing software.
Finally as promised. Code to extract significant cluster from whole brain voxel-wise statistics, in this case from FSL's Randomise output:
Extract binary files for each significant clusters:
#Extract Cluster values for all significant maps
dir=/Volumes/Vol/processed_DTI/
cd $dir;
rm modality_index.txt;
for study in DTI/STUDY_randomise_out DTI/STUDY_randomise_out2;
do
prefix=`echo $study|awk 'BEGIN{FS="randomise_"}{print $2}'`;
cluster -i ${study}_tfce_corrp_tstat1 -t 0.95 -c ${study}_tstat1 --oindex=${study}_cluster_index1;
cluster -i ${study}_tfce_corrp_tstat2 -t 0.95 -c ${study}_tstat2 --oindex=${study}_cluster_index2;
num1=`fslstats ${study}_cluster_index1.nii.gz -R|awk 'BEGIN{FS=""}{print $2}'|awk 'BEGIN{FS="."}{print $1}'`;
num2=`fslstats ${study}_cluster_index2.nii.gz -R|awk 'BEGIN{FS=""}{print $2}'|awk 'BEGIN{FS="."}{print $1}'`
echo $prefix"," $num1 "," $num2;
echo $prefix"," $num1 "," $num2>>modality_index.txt;
#loop through significant clusters
count=1
while [ $count -le $num1 ];
do
fslmaths ${study}_cluster_index1.nii.gz -thr $count -uthr $count -bin /Volumes/Vol/processed_DTI/STRUCTURES/${prefix}_${count}_neg.nii.gz;
let count=count+1
done
count=1
while [ $count -le $num2 ];
do
fslmaths ${study}_cluster_index2.nii.gz -thr $count -uthr $count -bin /Volumes/Vol/processed_DTI/STRUCTURES/${prefix}_${count}_pos.nii.gz;
let count=count+1
done
done
Extract cluster means:
#Extract TFCE cluster means
rm -f *_weighted.nii.gz
rm -f *_weighted_output.txt;
rm -f *_merge.txt;
cd /Volumes/Vol/processed_DTI;
for i in DTI/STUDY_randomise_out DTI/STUDY_randomise_out2;
do
prefix=`echo ${i}|awk 'BEGIN{FS="/"}{print $1}'`;
suffix=`echo ${i}|awk 'BEGIN{FS="/"}{print $2}'`;
rm structures_all.txt;
cd /Volumes/Vol/processed_DTI/STRUCTURES;
for j in `ls ${prefix}_${suffix}*`;
do
pre=`echo ${j}|awk 'BEGIN{FS=".nii"}{print $1}'`;
echo $pre >> /Volumes/Vol/processed_DTI/structures_all.txt;
done
cd /Volumes/Vol5/processed_DTI/NOMOM2/TEMPLATE/normalize_2017;
rm scalars_all.txt;
echo $suffix > scalars_all.txt;
sh ./weighted_mean.sh /Volumes/Vol/processed_DTI/ structures_all.txt scalars_all.txt /Volumes/Vol/processed_DTI/${prefix}/${suffix} S01 S02 S03 S04;
done
Finally, just run the code to organize the output data.







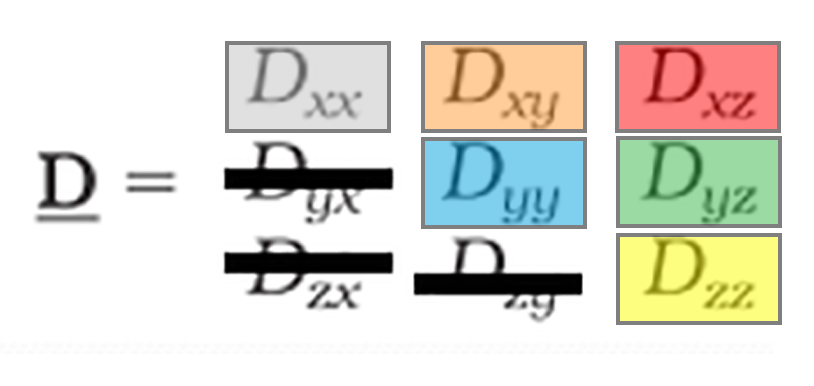































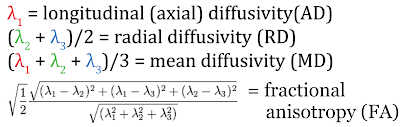













 and after (B) application of a real-time motion-correction algorithm. (MGH)")
